Zwei Typen von Fragen können (unter anderen) gestellt werden:
Zu welchem Individuum gehört eine DNA-Probe?
Die Genome verschiedener Individuen sind weitgehend identisch. Man schätzt jedoch, dass auf 300 bis 1000 bp ein Basenpaar nicht identisch ist. Für das ganze Genom ergibt das ca. drei Millionen "individuelle Basenpaare". Untersuchungen haben gezeigt, dass die Unterschiede nicht gleichmässig über das ganze Genom verteilt sind. Es sind bestimmte Regionen der DNA, vor allem repetierte Sequenzen, welche in einzelnen Individuen unterschiedlich sind. Mittels Restriktionsverdauung von DNA und Southern Blotting mit einer radioaktiven DNA-Sonde für die entsprechende repetierte Sequenz, können die Unterschiede sichtbar gemacht werden: RFLP = Restriktionsfragment-Längenpolymorphismus. Mittels PCR können selbst kleinste Mengen von DNA (z.B. aus einem Haar) so stark amplifiziert werden, dass sie für eine RFLP- oder SSCP-Analyse genügen.
Anwendung: Gerichtsmedizin: Spurenanalyse, Vaterschaftsanalyse
|
|
Im Atelier gibt es zu diesem Thema ein Video mit dem Titel "Genkrimi". |
Ist ein bestimmtes Gen in einer DNA-Probe verändert?
Bei dieser Fragestellung geht es darum, ein Gen oder eine Region eines Gens für einen bekannten Defekt (Punktmutation, Deletion, Addition) abzusuchen.
Anwendung: Pränatale Diagnostik: Erbkrankheiten
Beispiele: Hämoglobin S, Muskeldystrophie, Cystische Fibrose
Postnatale Diagnostik: Erbkrankheiten. In der postnatalen Diagnostik kann auch die Frage gestellt werden, ob fremde DNA in der DNA-Probe vorhanden ist, z.B. bei bakteriellen und viralen Infektionen.
Weil das humane Genom sequenziert ist, wird die prä- und postnatale Diagnostik ständig erweitert werden.
|
|
Sie können sich ein Beispiel für die Diagnose einer Infektionskrankheit im Atelier |
|
|
|
|
Unter pränataler Diagnostik versteht man die vorgeburtliche Erfassung von Erbkrankheiten.
Aus der ursprünglichen Schwangerschaftsvorsorge ist in den letzten Jahren eine eigenständige medizinische Disziplin entstanden, die sich um das Wohlergehen des werdenden Kindes bemüht. Die pränatale Diagnostik verwendet nicht-invasive und invasive Methoden.
Nicht-invasive Methoden:
Zu den nicht invasiven Methoden zählt die Ultraschalldiagnostik. Sie hat ihre Bedeutung nicht nur im Hinblick auf das Erfassen möglicher Missbildungen, sondern gilt heute als Standardverfahren zur Schwangerschaftsüberwachung. Sie liefert neben wichtigen geburtshilflichen Daten immer auch Informationen über mögliche foetale Missbildungen. Schwerere Missbildungen können mit Hilfe der Ultraschalltechnik in der frühen Schwangerschaft erfasst werden. So zum Beispiel Anencephalus (fehlende Grosshirnanlage), Spina bifida (offener Röcken), Zwerchfellmissbildungen, Herzfehler. In der späten Schwangerschaft sind sogar Defekte, wie zum Beispiel die Lippen-Kiefer-Gaumen-Spalte oder das Down-Syndrom, feststellbar.
Invasive Methoden:
Beide Methoden, die Amniozentese und die Chorionbiopsie, haben je nach Quellen ein Abortrisiko von 1-3%.
|
|
Im Atelier können Sie sich das Video Amniozentese und Chorionbiopsie ansehen. |
Die wichtigsten Angaben zur Muskeldystrophie Duchenne:
Klinik: Schwere, progredient verlaufende Muskelerkrankung. Die Duchenne-Muskeldystrophie (DMD) und die wesentlich mildere Verlaufsform nach Becker (BMD) entstehen durch Mutationen des Muskelproteins Dystrophin, dessen Gen auf dem X-Chromosom lokalisiert ist. Erste Symptome mit drei bis fünf Jahren. Rollstuhlabhängigkeit mit ca. zwölf Jahren. Lebenserwartung kaum mehr als zwanzig Jahre. Zur Zeit noch keine kausale Therapie. Häufigkeit: 1:3500 (männliche Geburten). Erbgang: geschlechtsgebundener, rezessiver Erbgang. Gen: auf X-Chromosom. 2,5 . 10 hoch 6 Basenpaare, grösstes zur Zeit bekanntes Gen mit 65 Exons. mRNA: ca. 14 000 Basen lang. Genprodukt: Dystrophin, Molekulargewicht 400 000 Dalton. Defekt: Deletion eines Teils des Gens. Der Unterschied zwischen BMD und DMD liegt darin, dass im Falle der DMD Deletionen im Dystrophin-Gen zu Verschiebungen des Leserasters und zum Kettenabbruch führen, während im Falle der BMD, trotz einer Deletion (z.B. eines ganzen Exons) der offene Leseraster des Gens korrekt weitergeführt wird: es resultiert ein Protein mit eingeschränkter Funktion.
Die Funktion des Dystrophins besteht nach gegenwärtigen Vorstellungen in einer Verankerung von Actin-Filamenten an Membranproteinen des Sarkolems. Der Verlust dieser Funktion scheint für die Degeneration des Muskelgewebes verantwortlich zu sein. Auch das Myocard (Herzmuskel) ist betroffen.
|
|
Im Atelier sind zum Thema "Progrediente Muskeldystrophien weitere Informationen und das Video "Duchenne Muskeldystrophie" aufgelegt. |
|
|
Zur Illustration der Diagnose "Duchenne Muskeldystrophie" können Sie einen Fall bearbeiten: |
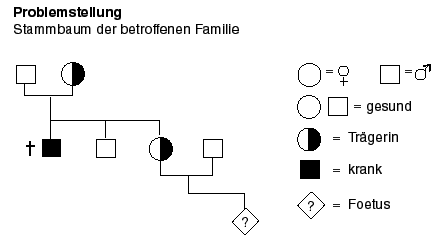
Hat das Genom des Foeten die Deletion, die zu DMD führen wird?
Vorgehen:
1. Entnahme von Zellen des Foeten: siehe Amniozentese oder Video "Amniozentese, Chorionzottenbiopsie"
2. DNA-Isolation: siehe "Chemie der Nukleinsäuren" und Video "DNA-Isolierung"
3. Schneiden mit Restriktionsenzymen
5. Hybridisierung:
|
|
Video-Clips: Hybridisierung im Labor |
Technische Hinweise | |||
|
. |
|
| |||
|
-> |
-> |
-> |
-> |
| |
Lösung der Problemstellung
Wichtigste Angaben zur Cystischen Fibrose:
Klinik: Funktionsstörung der exokrinen mukösen Drüsen. Lunge: chronische Bronchitiden, rezidivierende Pneumonien. Verdauungsorgane: Pankreasinsuffizienz führt zu Maldigestionen und Malabsorption. Meconium-Ileus, Diabetes mellitus, Leberzirrhose. Häufigkeit: Häufigste Erbkrankheit mit letalem Ausgang in der kaukasischen Bevölkerung, 1 auf 2000 Lebendgeburten, 1 auf 21 Personen ist Träger der Mutation (ca. 5% der Bevölkerung). Erbgang: autosomal rezessiv. Gen: liegt auf Chromosom 7; 27 Exons, 250 000 Basenpaare. Genprodukt: CFTR (Cystic fibrosis transmembrane conductance regulator) = Chlorid-Kanal in der Zellmembran. Defekt: Deletion eines Basentripletts in Exon 10 (häufigste Mutation; insgesamt etwa 180 Mutationen bekannt)
|
|
Im Atelier sind weitere Dokumente zu diesem Thema und das
Video "Im Sommer sterb ich nicht so leicht" aufgelegt. |
|
|
Zur Illustration der Diagnose "Cystische Fibrose" können Sie einen Fall bearbeiten: |
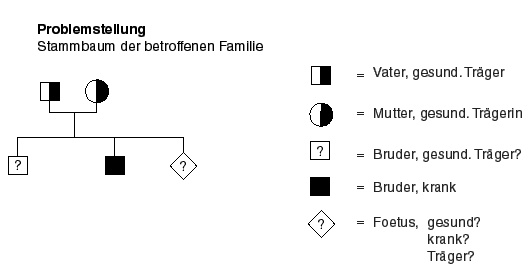
a) Ist der Foetus gesund oder krank?
b) Ist der gesunde Bruder des Patienten Träger des Gens?
Vorgehen:
|
|
Video-Clip: |
Technische Hinweise | ||
|
|
Video-Clip: |
Technische Hinweise | |
|
|
Video-Clip: |
Technische Hinweise | |||
Lösung der Problemstellung
|
|
|
|
Gentechnologie beim Menschen |
somatische Gentherapie |
